Nature Biomedical Engineering: XTIPC Research Progress on AI-powered Precision Oncology
Editor: HU Lun | Jan 13,2025
According to the World Health Organization, the number of cancer patients worldwide is steadily increasing, highlighting cancer as a major challenge that severely threatens human health. The prevention and treatment of cancer have thus become a global priority. Identifying cancer-driver genes plays a crucial role in understanding the mechanisms underlying cancer development, paving the way for personalized and precision therapies. However, existing methods still face significant challenges in terms of generalizability and interpretability.
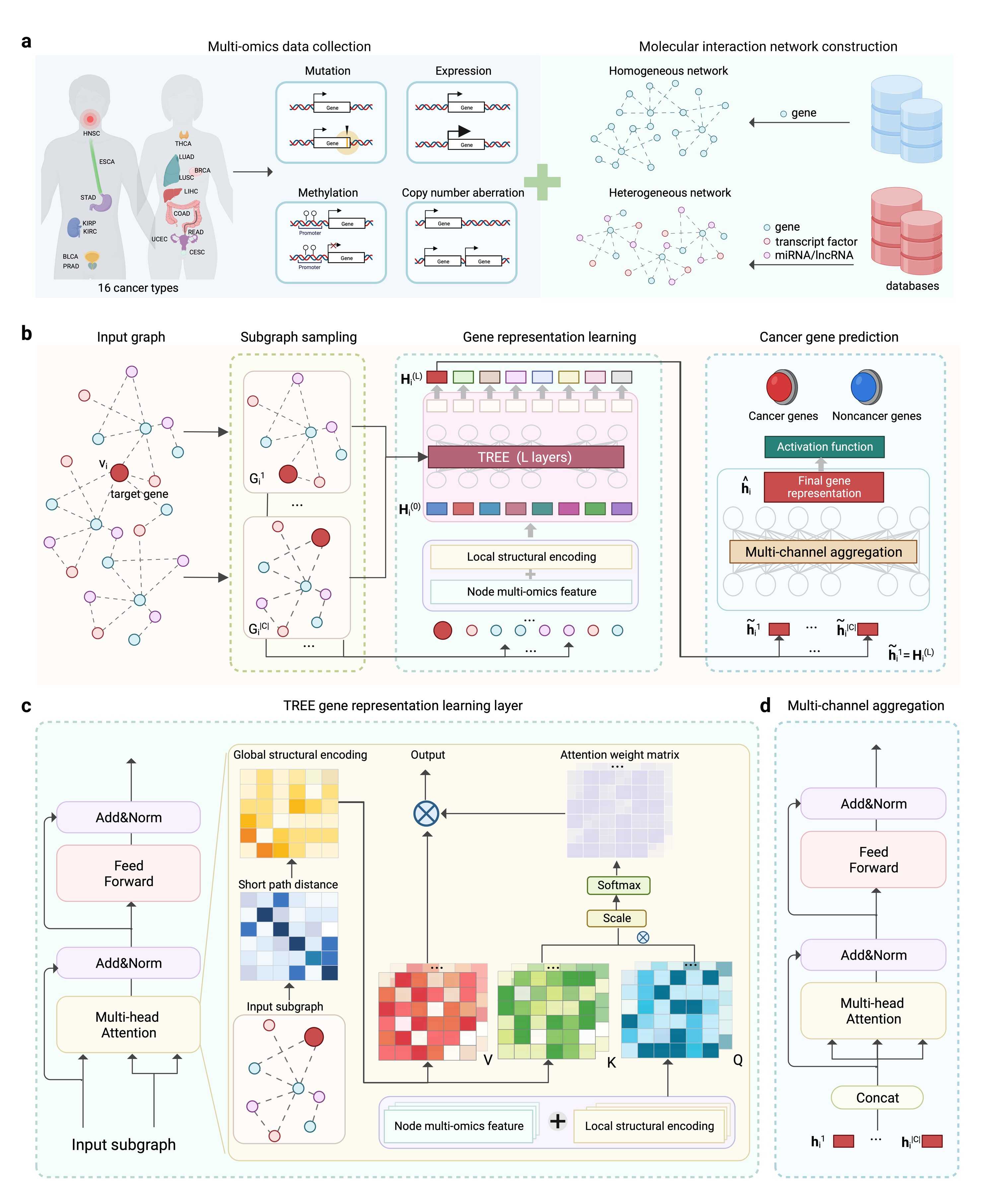
To address this issue, the research team at the Xinjiang Institute of Physics and Chemistry, Chinese Academy of Sciences, in collaboration with other experts, proposed a graph machine learning model, namely TREE, based on the Transformer framework. With this novel Transformer-based architecture, TREE not only identifies the most influential omics type but also detects the most representative network paths involved in regulating genes that drive cancer formation and progression. Notably, instead of training TREE on the complete, large input graph, we propose training it on subgraphs sampled from local structures. This approach enables efficient node-level representation learning while significantly reducing computational resource requirements. Unlike traditional Transformer architectures, TREE incorporates graph structural information from biological networks as part of its input. It further integrates position embeddings, derived from node degree information, with the multi-omics features of nodes in the input layer. Moreover, during the modeling of graph spatial structures, TREE employs a co-attention mechanism, where global structural encodings of nodes, learned from network distance information, guide the calculation of attention weights. This design enhances the model's ability to capture complex relationships within biological systems.
Hence, TREE integrates the strengths of artificial intelligence and biomedicine, delivering not only reliable predictive performance but also enhanced generalization and interpretability. By incorporating multi-omics data from genes and other biological molecules along with structural information from both homogeneous and heterogeneous biological networks, the model significantly improves the accuracy of cancer driver gene predictions. This advancement enables more precise identification of genes closely associated with cancer progression, laying a solid scientific foundation for the development of personalized treatment strategies. Additionally, the model markedly enhances the efficiency of cancer driver gene identification, facilitating early cancer diagnosis and treatment planning. The model also excels in analyzing high-order associations within multi-omics data and network structures, which bolsters its interpretability. This feature provides researchers with invaluable insights into the regulatory mechanisms of cancer-related genes, allowing for the discovery of potential cancer driver genes. These insights hold profound implications for unveiling the mechanisms underlying cancer development and devising novel therapeutic strategies. By constructing a precise regulatory map of cancer genes, the model paves the way for advancements in personalized medicine and the development of targeted drugs. Moreover, the model's strengths in integrating multi-omics data and complex network analysis endow it with cross-disease and cross-disciplinary applicability. This research exemplifies the cutting-edge integration of artificial intelligence with biomedical engineering, offering innovative solutions to address the challenges of cancer. It marks a significant step forward in advancing life sciences towards a more precise and intelligent era.
This work has been published in full in the prestigious journal Nature Biomedical Engineering. The Xinjiang Institute of Physics and Chemistry, Chinese Academy of Sciences, is the sole corresponding institution. This work was supported by the National Natural Science Foundation of China, the Chinese Academy of Sciences, and Xinjiang Uygur Autonomous Region.
附件下载:
 86-991-3835823
86-991-3835823 lhszhb@ms.xjb.ac.cn
lhszhb@ms.xjb.ac.cn 86-991-3838957
86-991-3838957 40-1 Beijing Nan Road, Urumqi, Xinjiang, 830011, China
40-1 Beijing Nan Road, Urumqi, Xinjiang, 830011, China


Copyright @ Xinjiang Techinical Institute of Physics and Chemistry, Chinese Academy of Sciences. All Rights Reserved
